Xinwen Zhang, Nora Haanaes, Berenice García Rodríguez, Giovanni Volpe, Janet Kumita and Daniel Midtvedt
Date: 11 March 2026
Time: 18:00-20:00
Place: Aula Medica, Karolinska Institute, Solna
Conference Protein Folding in Real Time, 11-13 March 2026, Stockholm, Sweden
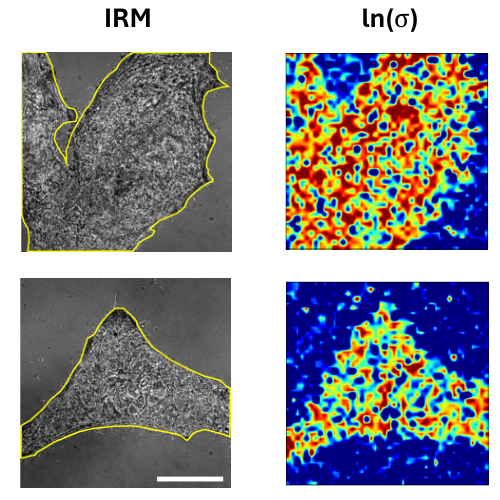
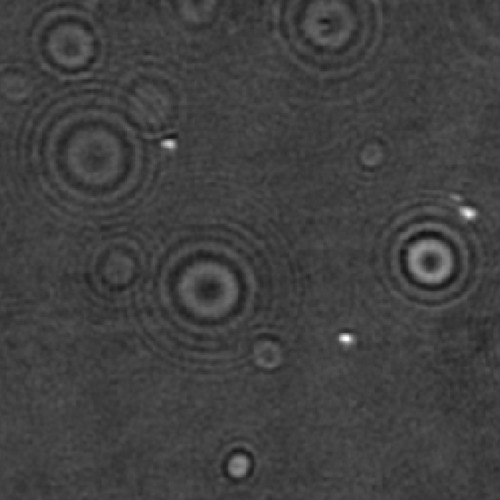
Biomolecular condensates formed through liquid–liquid phase separation (LLPS) play important roles in cellular organization, yet quantitative and label-free characterization of their physical properties remains challenging. In this work, we apply off-axis holographic microscopy to study a synthetic biomolecular condensate platform based on the LCD2-CTPR protein system. These proteins, composed of modular consensus-designed tetratricopeptide repeat (CTPR) domains fused to intrinsically disordered regions, undergo phase separation under varying salt concentrations. By incorporating short binding motifs such as ATG13 or Func1, the condensates can specifically recruit the autophagy-related protein LC3. Using label-free quantitative phase measurements, we analyze changes in condensate optical radius and refractive index during LC3 recruitment and over time. Our results show measurable variations in condensate size and optical properties, highlighting the sensitivity of these systems to compositional changes. This work demonstrates the applicability of holographic microscopy for quantitative characterization of synthetic biomolecular condensates and provides a framework for studying protein phase separation in a non-invasive manner.