Mite Mijalkov, Dániel Veréb, Oveis Jamialahmadi, Anna Canal-Garcia, Emiliano Gómez-Ruiz, Didac Vidal-Piñeiro, Stefano Romeo, Giovanni Volpe, Joana B. Pereira
Network Neuroscience 1-40 (2022)
doi: 10.1162/netn_a_00286
medRxiv: 10.1101/2022.03.08.22272089
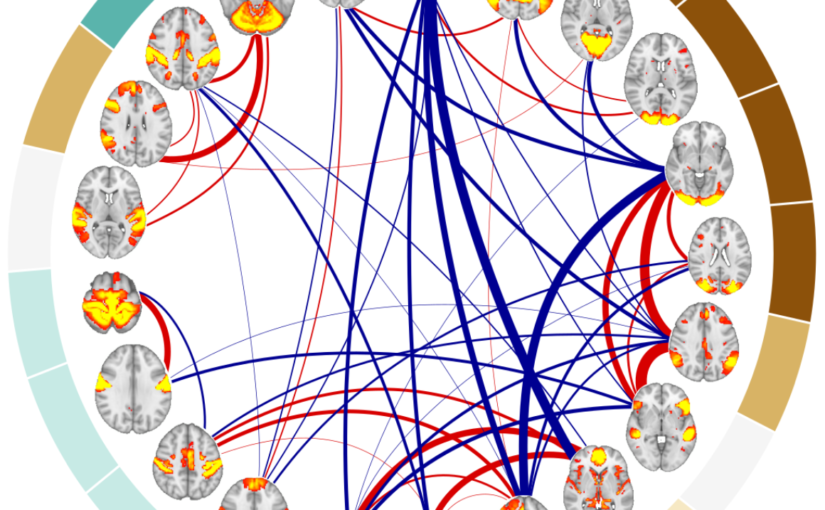
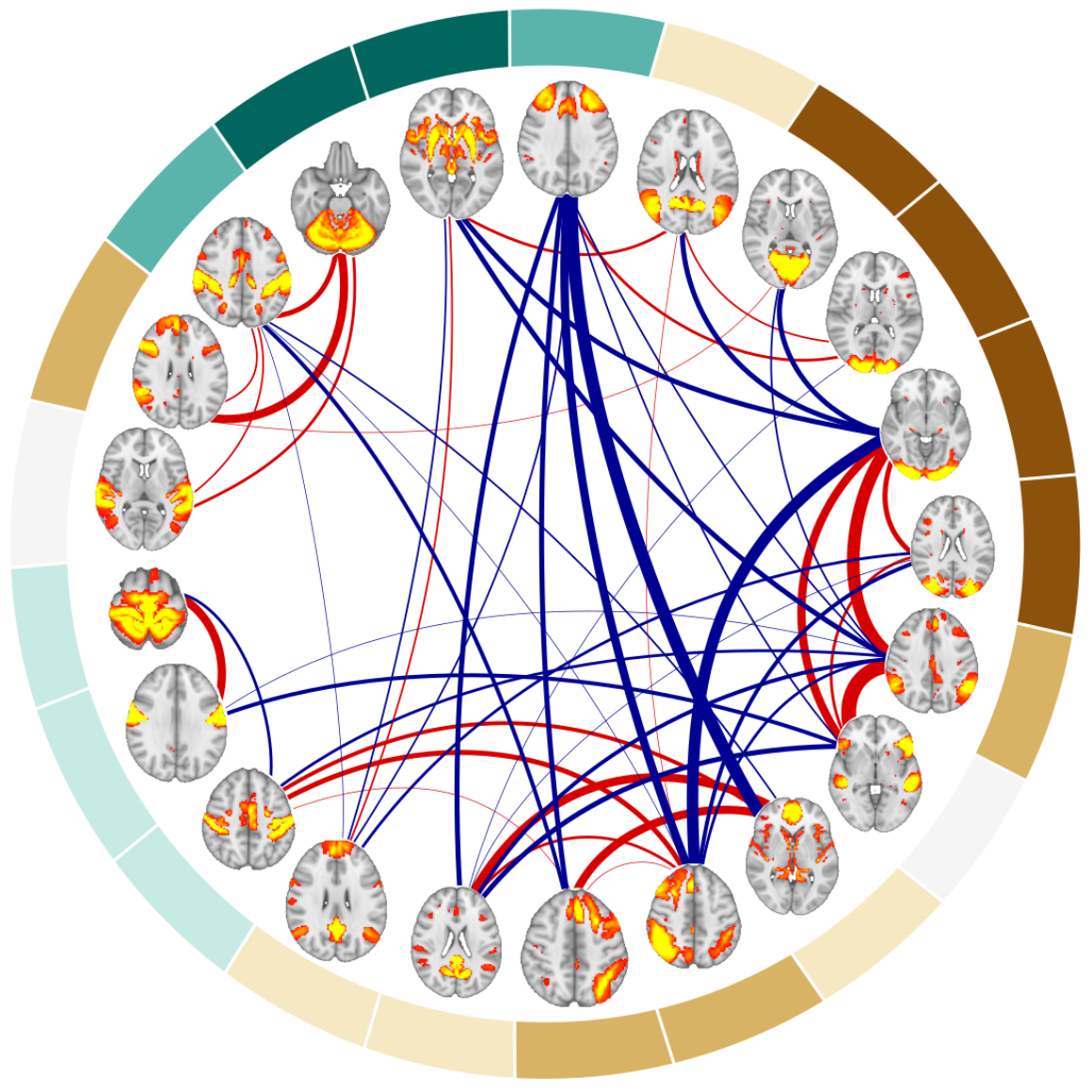
Aging is a major risk factor for cardiovascular and neurodegenerative disorders, with considerable societal and economic implications. Healthy aging is accompanied by changes in functional connectivity between and within resting-state functional networks, which have been associated with cognitive decline. However, there is no consensus on the impact of sex on these age-related functional trajectories. Here, we show that multilayer measures provide crucial information on the interaction between sex and age on network topology, allowing for better assessment of cognitive, structural, and cardiovascular risk factors that have been shown to differ between men and women, as well as providing additional insights into the genetic influences on changes in functional connectivity that occur during aging. In a large cross-sectional sample of 37543 individuals from the UK Biobank cohort, we demonstrate that such multilayer measures that capture the relationship between positive and negative connections are more sensitive to sex-related changes in the whole-brain connectivity patterns and their topological architecture throughout aging, when compared to standard connectivity and topological measures. Our findings indicate that multilayer measures contain previously unknown information on the relationship between sex and age, which opens up new avenues for research into functional brain connectivity in aging.